About Cordax
Cordax predictions rely on a rigorously annotated dataset of amyloid fibril core structural topologies. The tool uses a hexapeptide moving window threading process to calculate stability energies which are fed into a logistic regression model that outputs an aggregation propensity profile and provides structural models of the identified aggregation prone regions.
__
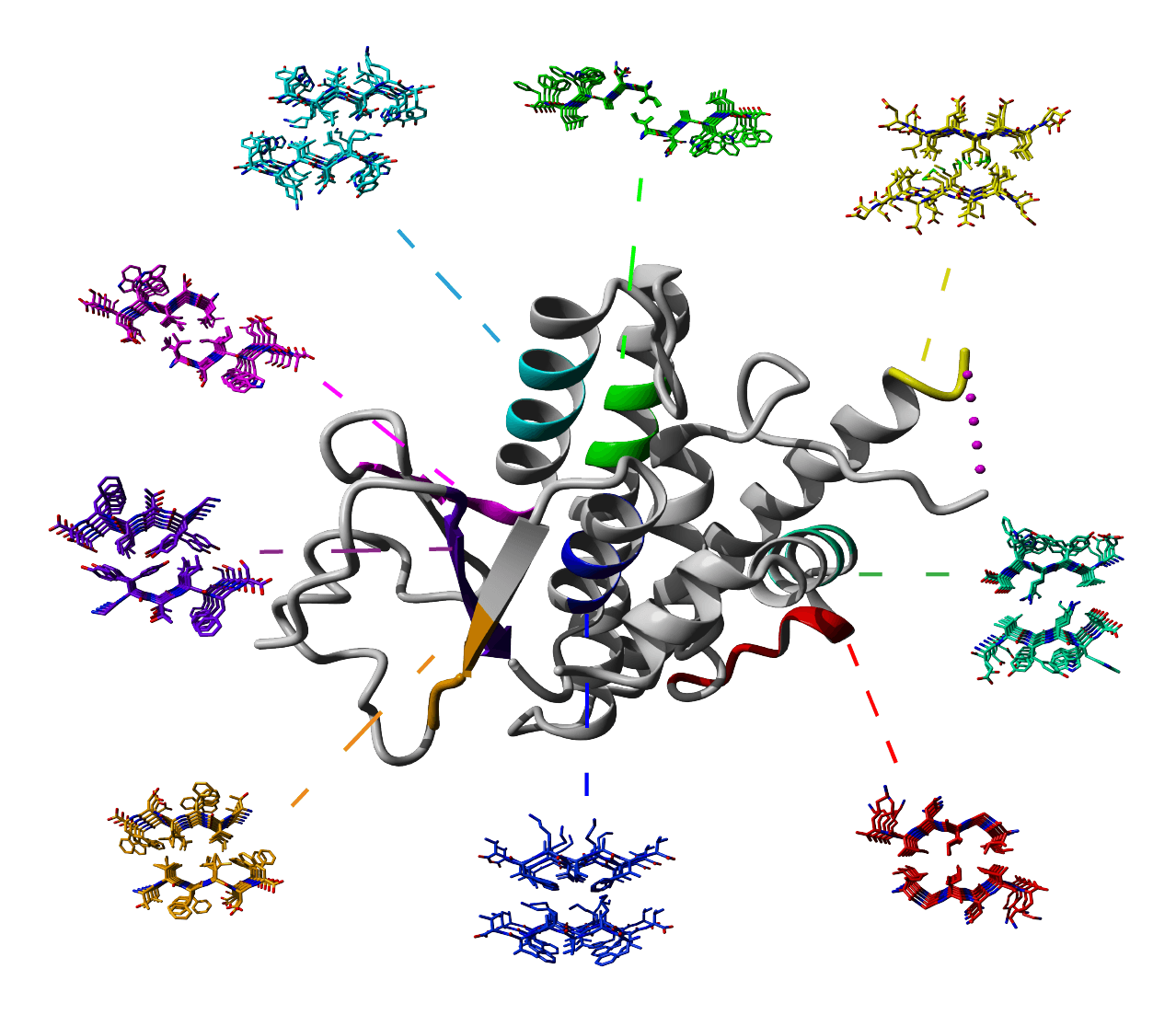
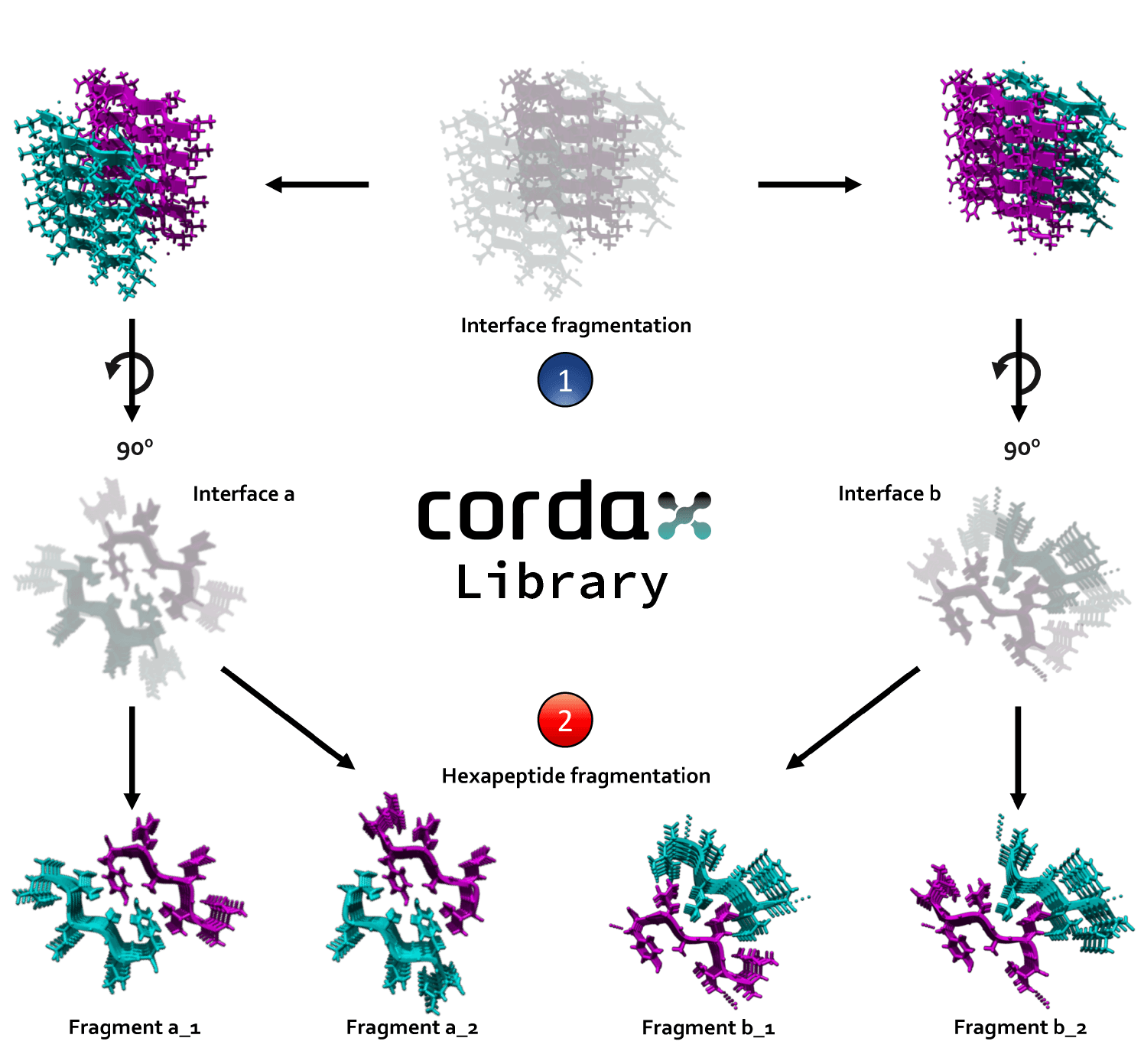
Cordax structural library
Cordax employs a structural library of 140 amyloid fibril core structures distributed in 7 distinct topological classes, as described by Sawaya et al. Nature 2007. Parallel architectures were split into separate individual entries when sharing more than one homotypic amyloid interfaces and the number of structural variants was expanded by breaking down longer segments into hexapeptide constituents.
__
Regression model training
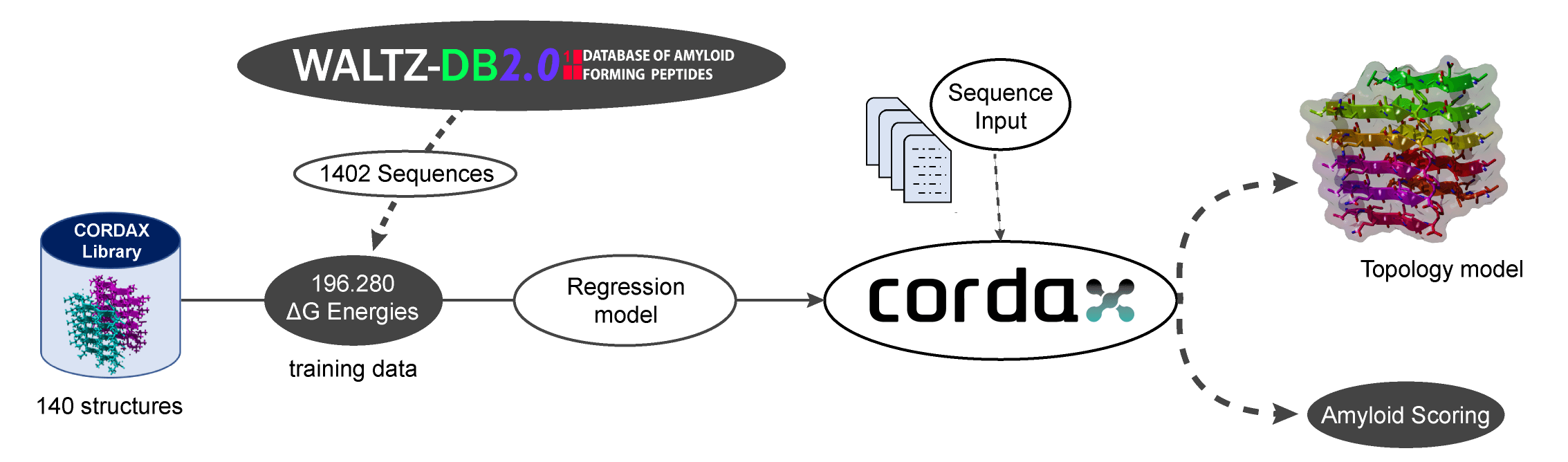
The regression model was trained against WALTZ-DB 2.0 (Louros et al. NAR 2019), the largest public comprehensive repository of experimentally defined amyloidogenic sequences derived from both functional and pathological amyloid-forming proteins. In total, 1402 hexapeptide sequences were modelled on the backbone structures of the Cordax library. The thermodynamic stability of each model (ΔG, kcal/mol) was fed into a logistic regression model to distil the aggregation propensity from the free energy values.
Data are transformed in a scoring range between 0 and 1, using a Min/Max scaling algorithm. The regression model was trained with L2 penality and regularisation strength (C) equal to 1. Both scaling of the estimated ΔG and the machine learning model were developed using the SciKit-learn python package.
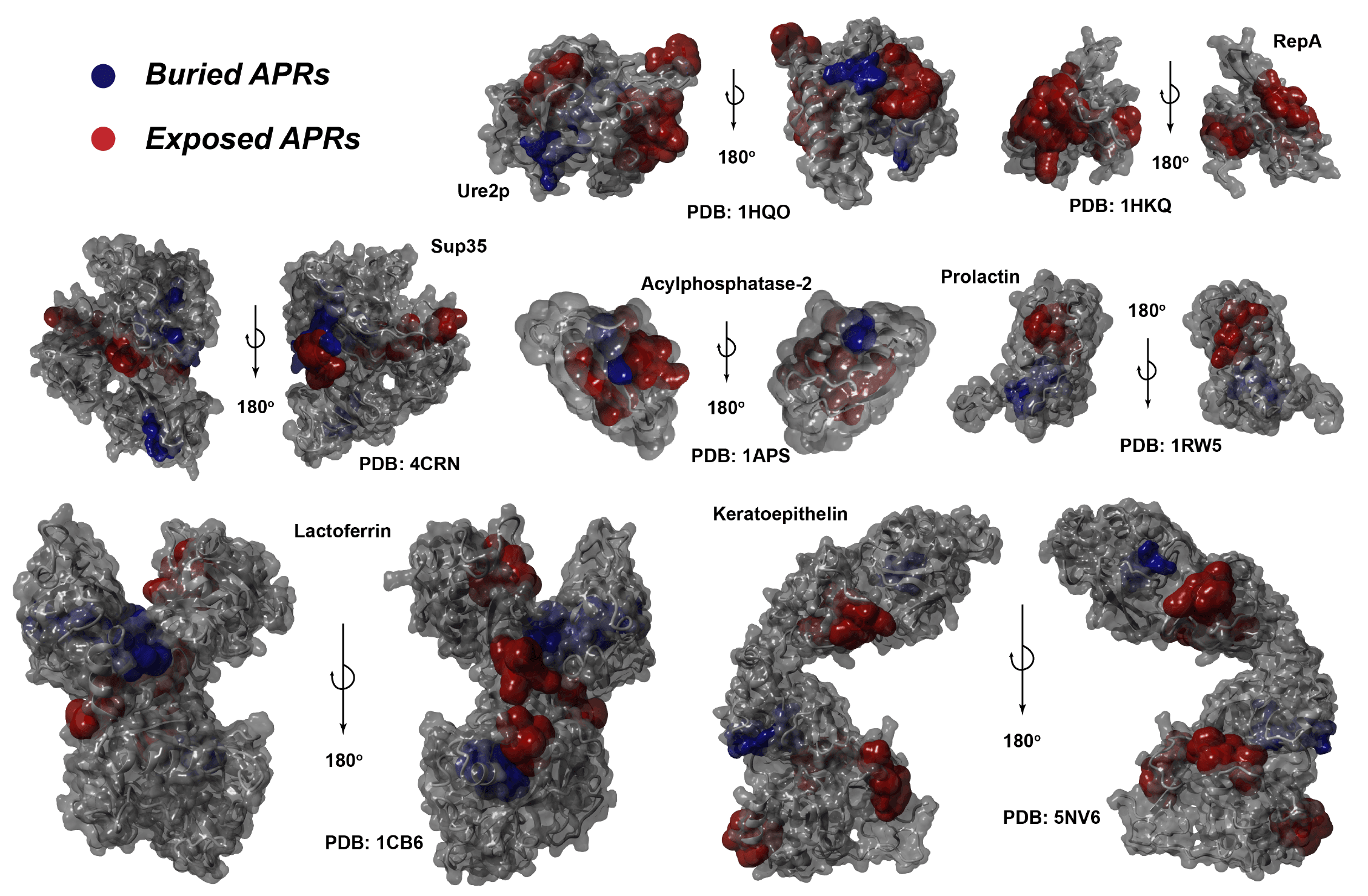
Cordax identifies soluble surface-exposed APRs
Cordax uses structural compatibility to predict aggregation prone regions (APRs) in proteins and as a result is unbiased towards by typical sequence propensities such as hydrophobicity or beta-propensity. This enables our method to discover new amyloid sequences sharing features such as:
- High solubility
- High net charge
- Surface exposure in protein native folds
- Composition similarity to phase transition sequences
- Disorder or α-helix propensity (conformational switches)
__
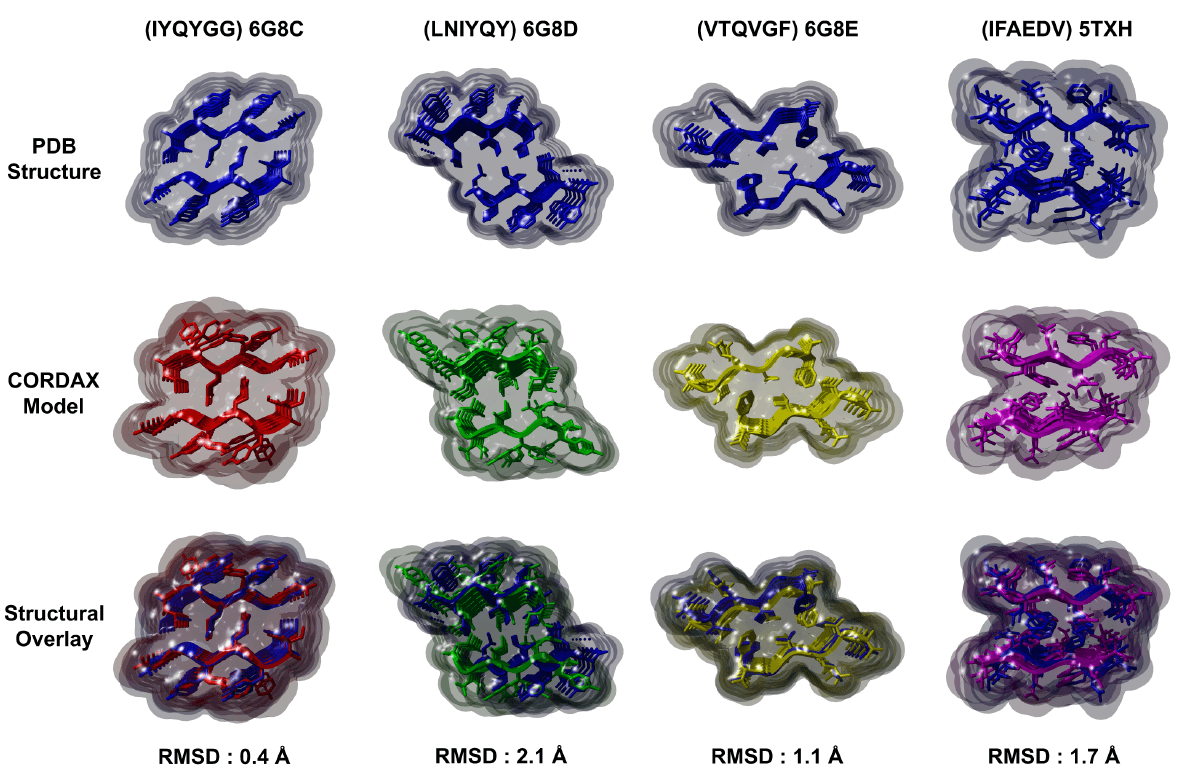
Prediction of fibril topologies
Cordax provides a cost-effective complementary powerful computational alternative to determine the structural layout of putative amyloid fibril-forming segments.
Every hexapeptide window of a protein sequence is cross-threaded against the Cordax library and energetic fits are evaluated using FoldX. Segments with scoring values equal or above the determined aggregation propensity threshold (0.71) are considered as aggregation prone regions and as a result, an energetically fitted structural model is outputed as the putative fibril topology.
******
References
- Louros, N. et al. (2020) et al. Structure-based machine-guided mapping of amyloid sequence space reveals uncharted sequence clusters with higher solubilities. Nat Commun 11, 3314 (2020).
- Louros, N. et al. (2019) WALTZ-DB 2.0: an updated database containing structural information of experimentally determined amyloid-forming peptides. Nucleic Acids Res, 48 (D1), D389–D393. https://doi.org/10.1093/nar/gkz758